Documentation
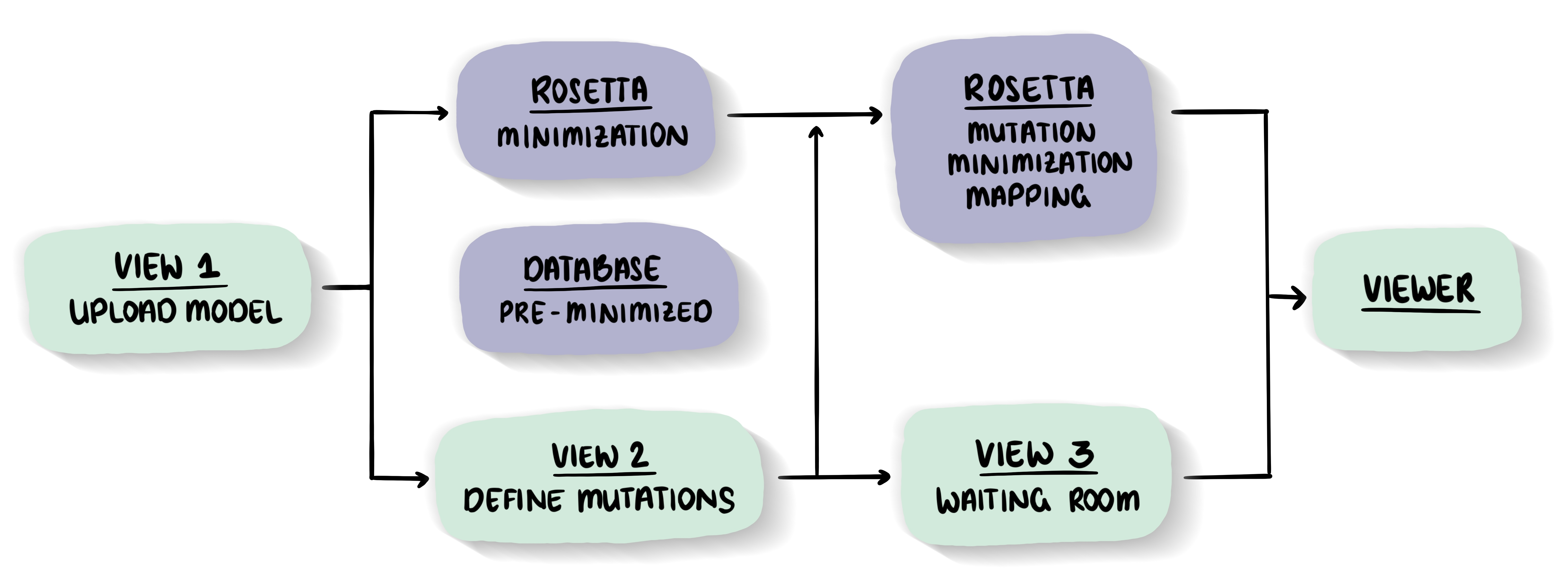
General philosophy and key elements of the MutationExplorer
-
MutationExplorer - exploring the effect of mutations on the structureThe MutationExplorer maps variants onto the protein 3D structure which allows to interactively explore the effects of mutations with respect to stability and function. Often wwPDB structures contains mutations and can not intuitively be mapped to its native sequence, sequences of structures changed to close or remote homologs or structures designed interactive while inspecting the structure. With the MutationExplorer this is now easily possible and even visualises the effect of the variant on the protein structures stability.
Structure file preparation
-
Energy minimization - the key to high qualityThe minimization is crucial for the quality of the outcome. The better the structure is energetically minimized, the more reliable the results will be.
We continuously pre-minimize structures from the PDB using the command-line described below. If you specify a PDB ID that is contained in our database, the pre-minimized structure is used.
If the used structure is not available in our database, we offer two modes of side-chain minimization: a short and a long one. The short one should take a few minutes, depending on the size of the protein. The long one may take some hours for large proteins. It is strongly recommended to use the long minimization, despite the significant waiting period. The minimization will start after submission of the mutations. There is also a notification if the structure has already been minimized. It is not possible to overwrite the minimization in the database.
-
AlphaFold and your own models
Structures from predictive tools such as AlphaFold and ESMfold are not pre-minimized and you should follow the steps below. Please note that the minimization has to be performed with the same energy functions than we use in the MutationExplorer, namely Rosetta.
You can use any of the following options: Either use one of the two structure minimization options we provide on the website or do it yourself via a local installation of Rosetta or via the ROSIE server.
-
Minimizations via the MutationExplorer
We offer a long and short side-chain only optimization.
For both minimizations, we use the Rosetta fixbb functionPATH_TO_ROSETTA/fixbb.static.linuxgccrelease -use_input_sc -resfile INPUT.res -nstruct 1 -linmem_ig 10 -in:file:s INPUT.pdb -out:pdb -out:prefix OUTDIR/The only difference between the long and the short optimization is the usage of more rotamers in the long optimization via
-ex1 -ex2Although the minimizations provided in the webserver are sufficient to get a first idea about mutation effects, we recommend you to perform a more through minimization, as described in the following. Please note that the structures within our database have been minimized using the more through approach below.
-
Do it yourself!
In the following, we suggest a more thorough minimization, which goes beyond a simple side-chain optimizations. It optimizes the backbone in a limited way (via the
-relax:constrain\_relax\_to\_start\_coordsoption). This does not allow a completely free backbone minimization as this might be leading to problems such as larger structural backbone permutations. Further, we recommend the more thorough relax function with a current energy score.PATH_TO_ROSETTA/relax.static.linuxgccrelease -relax:fast true -relax:cartesian true -score:weights ref2015_cart -use_input_sc -optimization::default_max_cycles 200 -linmem_ig 10 -relax:constrain_relax_to_start_coords -ex1 -ex2 -nstruct 20 -in:file:s INPUT.pdb -out:pdb -out:prefix OUTDIR/This commandline will create 20 models, from which you should select the one with the lowest energy. Each PDB contains a line starting with pose. In this line you want the last value, which is the total energy. This resulting structure can then directly be used in the MutationExplorer without requirement of further minimization.
-
ROSIE server
There is a way to perform the structure minimization without a local Rosetta installation by using the ROSIE 2 server (https://r2.graylab.jhu.edu/). It requires that you have a GitHub account with your academic email address linked and verified. Once you are logged in, you can select Rosetta from the Rosie apps list. There, you need to upload your PDB structure and paste the following into the Rosetta flags file field:
-in:file:s input.pdb -in:file:fullatom -constrain_relax_to_start_coords -relax:ramp_constraints false -nstruct 6 -out:suffix _Relaxed #rosie type serial #rosie tasks 6The job will run for about 2-4 hours, depending on the protein size. Once it is done, you can go to job files and find score\_Relaxed.sc. You can open it on the website, if you do so, you need to select raw in the view as options. In the total score column, find the row with the lowest energy score and look up the name in the description column. Then download the input\_relaxed .pdb file with the respective number. This is the file you are going to use as an input to the MutationExplorer.
-
Database of pre-minimized structures from the Protein Databank (PDB)
Since the minimization is essential for correctly highlighting the energetic impact of mutations, but takes a substantial amount of time to perform, we calculated a database of pre-minimized structures from the PDB. Currently our database contains 45,000 models. You will be notified if your PDB ID was found in our database or has to be calculated.
Mutational tools - under he the hood of MutationExplorer
-
RaSP - bringing exploration to full bloom
RaSP is a new deep-learning-based tool that rapidly estimates protein stability changes. RaSP predictions strongly correlate to scores from Rosetta calculations which demand longer compute times.
With RaSP, MutationExplorer presents the user with a quick initial estimation of a mutation’s (de)stabilizing effect, without having to wait for the longer full minimization process.
RaSP will calculate all mutations for all amino acids in a chain within ~10 minutes.
If this option is selected, you can press the Ctrl key and left-click on the residue in the viewer. This will open a frame with 20 energy estimates. Be aware that this is an approximation but can be used for further guidance and selection of amino acids.
Interpretation of the results
-
Interpretation of the results
The more negative a residue's energy is, the more stabilizing its effect onto the structure. Similarly, mutations that lower the energy are considered stabilising. Within the viewer, we annotate stabilizing mutations or residues in blue, while destabilizing residues are highlighted in red. As a general rule, exposed residue such as the very N- or C-terminus are often very flexible and therefore structure-based stability calculations may not be suitable in those regions.
Depending on the design goal, it is worth keeping in mind that flexibility is often required for function, and hence optimization towards completely rigid structures might cause problems to the function of the protein. For scaffold proteins where the main function is structure or stability, this is not an issue.
Getting started with MutationExplorer
-
Manual mutation syntax
Define a list of point amino acid substitutions with the following syntax:
- A comma separated list of:
CHAIN : RESIDUE-ID TARGET-AA-TYPE - Example:
A:23G,B:123A
This mutates the residue in chain A with the PDB residue ID 23 to glycine and in chain B the residue with residue ID 123 to alanine.
Note that the all mutations will be done in the same model, multiple individual mutations are best done via the explorer interface
- A comma separated list of:
-
Sequence AlignmentYou can mutate the model that you uploaded in previous step using a sequence alignment in ClustalW format. One of the sequences in the alignment has to match exactly one sequence in the PDB or be a subsequence of it that you uploaded in previous step. Since one alignment is associated with one chain, you can upload up to three alignments. If you need to mutate more than three chains, you can upload target sequences for the other chains or define mutations manually.
-
Target Sequence
Provide the sequence for each chain of the uploaded PDB that it will be mutated to. The target chain sequences as a chain of one-letter sequences can be provided as FASTA files (top), pasted directly (center) or directly fetched from UniProtKB (bottom). For each sequence, select the corresponding PDB chain from the dropdown menu. You can also combine different uploads for multi-chain proteins (e.g. Fasta for Chain A, Paste sequence for Chain B). Please ensure that the uploaded/pasted sequences exactly match the sequence in length (check your PDB!) and avoid sequence offsets resulting in mutation of all residues.
-
Using VCF files (sequencing data)
VCF, the tab-delimited Variant Call Format, is incredibly powerful and flexible. As example of the minimal input format used to upload SNPs to MutationExplorer, see the sample file RTEL1.vcf, described in our manuscript. This tutorial lists the steps you will take to upload RTEL1.vcf and replicate the example page RTEL1 MutationExplorer results.
Step-by-step guide
- From the home page, click on upload vcf and you will see a new form with a detailed explanation of MutationExplorer VCF workflow and limitations.
- Select long minimization to replicate the energy values of our example page. Or, quick is OK if numerical precision is not a concern. None is fastest, but energetic calculations will be meaningless. Click the i at right for more details.
- Click the check under “RaSP” if you wish to skip this stability prediction calculation. Click the i at right for more details.
- Click submit to start the calculations. The webpage tab at the top of the browser window
will immedately indicate a refresh in progress. Very soon you are transported to a new page
showing Status of Job
nnnnn. Be patient as the calculation proceeds. It is a good idea to right-click on Result Page for later references. However, this URL will not be active until the various calculations complete. At that time, you will be automatically redirected to this same Result Page. Your patience will be well rewarded.
Troubleshooting your VCF uploads
The processes between the submission of your VCF file, and the display of an AlphaFold model are complex. Your VCF upload will be processed by the Ensembl Variant Effect Predictor (VEP), running locally on the MutationExplorer server. If your results are unexpected, we recommend you paste your VCF file into the public VEP website: https://ensembl.org/Tools/VEP. This is the fastest way to ensure that your VCF file is properly tab-delimited and contains the minimum required columns and headers. The raw output from the website should also list protein missense variants.While seeing missense variants from the public VEP website is encouraging, other difficulties may remain. For example:
- MutationExplorer can only analyze one protein at a time. You will need to split your VCF file and make separate runs, if your VCF file generates missense variations for multiple genes.
- The VEP often returns Ensembl transcript Ids which do not map to Swiss-Prot curated, canonical UniProt transcript IDs. Without a UniProt ID, MutationExplorer cannot retrieve an AlphaFold model. You may manually check cross-references between ENST… transcript IDs and Uniprot IDs at ensembl.org and uniprot.org, respectively.
- Inputting GRCh37/hg19 genomic positions, which are indistinguishably formatted, will result in wildly incorrect results. You must first “liftover” such coordinates, by converting your VCF coordinates to BED format, then submitting them to https://genome.ucsc.edu/cgi-bin/hgLiftOver. As you use these kinds of tools, remember that MutationExplorer requires GRCh38.
The main stage: MutationExplorer result window
-
General
In the explorer view, you will be presented with information about your set of mutations, relative per-residue energies and visualization of the protein.
Left side (info bar):
- Select
- Tree View: Outlines the mutations that were performed, where mut_0 will be the unmutated, minimized PDB file that serves as the basis of mutations. You can select entries in the tree view for individual mutations.
- Color by Dropdown menu: provides different coloration methods (absolute energy, energy difference to parent, absolute hydrophobicity, difference in hydrophobicity to parent, conservation, absolute interface score, difference in interface score to parent) for the coloration of the protein in the main window.
- Download all button: pack all PDB files into a ZIP archive for downloading.
- Mutate
- Enter mutations field: allows for further mutations to be entered, with the root being the currently active protein (printed in bold in the tree view). The "info" field onm the bottom provides info on the currently selected protein.
-
Info
- Table Showing information about your current selection: Total Energy (Rosetta Energy units), the parent structure, introduced mutations and the exploration ID.
Right side (main window):
- Mol* visualization of the protein with a blue-white-red color gradient, where blue represents negative values and red positive values in the default color setting. The mutated residues are highlighted with a red translucent sphere. Hovering over the cartoon will highlight residues and display a tooltip with further information in the bottom right corner.
- MDSrv Window: By default, shows a sequence alignment of your root and mutated protein with the currently mutated residue highlighted in red. Hovering over cartoon residues will also highlight the residue in the alignment and vice-versa. It will further display a tool-tip on the right side with information about the highlighted residue.
Clicking on any highlighted residue will zoom in, showing the selected residue and its surroundings in a ball-and-stick view. Different carbon colors indicate protein chains, otherwise atoms are colors by type. Dashed lines indicate residue-residue interactions like hydrogen bonds.
- Select
-
Adding mutations
- In the tree view, click on the protein to serve as the root for your next mutation, e.g. mut_0_1, which is the first mutation entered in the webpage.
- In the Mutate field, enter the mutations to be added to the protein. Click on the info box for more informations.
- Click on submit and wait for your new protein to be added. The waiting message shows the name of the new mutation. In the tree, the new mutated protein will appear indented under the selected root.
-
Adjusting visualization
You can adjust the metric that serves as the basis for coloration in the \textit{"Color by:"} Dropdown menu. By default, the colormap will be from -2 to 2 in a blue-white-red gradient
- Click on the Change Visualization tab in the bottom right of the main window to adjust the color scale range and the mutation markers.
- First select for which structure you want to change the range of the color scale by choosing the corresponding file in the Structure drop-down menu. On the left and right side of the color scale, change the numbers and confirm with enter. The color scale will be adjusted upon clicking Apply below the scale.
- The mutation marker color and size can be adjusted via the slider and color window. The marker can be turned off by clicking on On in the Show Field, setting it to Off.
- It is possible to hide all mutation spheres at once. Click the Disable all mutation spheres button in the Mutation Markers panel. To show all mutation spheres again, click the Enable all mutation spheres button.
- If you are importing multiple structures into a scene, it may be beneficial to hide a specific structure. Select the structure you want to hide from the Structure drop-down menu. Then click the Hide Structure button at the bottom. To make the structure visible again, simply click the Show Structure button.
Further applications
-
Homology modeling of multiple states of a protein
What AlphaFold is not able to do is to model different states of a protein. This can be achieved using MutationExplorer. For many classes and families of proteins, multiple states are available in the PDB. Different states can be modeled By selecting different PDBs as base structure and follow these steps:
- Under upload structure and mutations enter the PDB ID of the base structure or upload a model from your machine.
- Ideally select the long minimization (for homology modeling the RaSP option can be disabled) and click the next button.
- For uploading the sequence to which you want to modify the base structure to there are multiple choices:
- Paste or upload it under target sequence (when the target is rather similar to your base structure / template).
- If your target sequence has an Uniprot ID you can enter that.
- You will have to indicate to which chain in the base structure the sequence shall be applied (available chains are listed).
- If it is a very remote relationship, follow the AlignMe procedure.
- For other cases either also use AlignMe, or upload an alignment from another source via “sequence alignment”.
The part that cannot be done by MutationExplorer is the minimization of the model. The user can choose between different possibilities, e.g. using Rosetta as explained above, or by performing MD simulations and many others. Whatever approach the user choosed, it requires sufficient sampling and thus has to be done locally.
-
Backmutating PDB structures to wildtype sequence for MD simulations
Structures deposited in the PDB are very often modified in order to solve them experimentally. Using MutationExplorer, it is a simple two-step process to mutate them back to their native sequence. Thus it is common step for simulating proteins to back-mutate PDB.
However, it should be noted that at its current state MutationExplorer is not able to complete the structure by modeling missing parts in the PDB.
-
Multi-state modeling of homologs via Molecular Dynamics simulation ensembles
Molecular dynamics simulation can generate ensembles of functionally relevant states. MutationExplorer maps those ensembles to close or distant homologs. For close homologs, an alignment will be automatically generated internally. Alternatively, a pre-calculated sequence alignment can be provided. Where more refined alignments are required, the server accepts results forwarded from the AlignMe website https://doi.org/10.1093/nar/gkac391. Such sequence alignments carry nuanced details and MutationExplorer drills down to reveal them all.
-
Aiding design for experimental studies for protein characterization
When designing proteins, the effect of the variant is often only known after applying of an external software, via multiple design rounds. After each of those, the structures can be inspected. The power of the MutationExplorer lies in the possibility to do this directly in the viewer and select novels rounds of mutations immediatley and due to their influence on the structure.
-
(De-) Stabilize a protein-protein interface
3D mapping of energies is not limited to monomers. Over single or multiple rounds of mutations, protein complexes or protein-protein interfaces can flexibly be designed to achieve (de)stabilization. The effect of each design choice can be explored visually.
Methods / code sources
-
Structure minimization
We use the following Rosetta code to minimize the structure. The basis is the fixbb function, while the resfile does not contain any indicated mutation.
PATH_TO_ROSETTA/fixbb.static.linuxgccrelease -use_input_sc -in:file:s path_to_structure -resfile path_to_empty_res_file -nstruct 1 -linmem_ig 10 -out:pdb -out:prefix prefix -ex1 -ex2 -
Mutation
We use the following Rosetta code to mutate the structure. The basis is the fixbb function.
PATH_TO_ROSETTA/fixbb.static.linuxgccrelease -use_input_sc -in:file:s path_to_structure -resfile path_to_empty_res_file -nstruct 1 -linmem_ig 10 -out:pdb -out:prefix prefix -
VCF processing
We process your VCF format file by submitting it, unmodified, to the ENSEMBL Variant Effect Predictor (VEP).
Our wrapper script:
vcf2rosetta.py VCF_fileinvokes vep with a command line that includes references to our local ENSEMBL cache files and these flags
vep -i your_input_file.vcf --format vcf --no_progress --buffer_size 100000 --check_existing --symbol --protein --uniprot --domains --canonical --biotype --coding_only --vcf --db_version 105 --assembly GRCh38 --no_stats -o stdoutFrom the extensive vep raw output, the wrapper script extracts revealed missense variants and impacted ENSEMBL transcript identifiers. For display in MutationExplorer, we retain only variants on transcripts which cross-reference to Swiss-Prot curated, canonical, Uniprot protein IDs, which which we retrieve alphafold models.
-
RaSP
Details about the RaSP software can be found here: https://doi.org/10.1101/2022.07.14.500157
The RaSP software code can be found on GitHub: https://github.com/KULL-Centre/papers/tree/main/2022/ML-ddG-Blaabjerg-et-al
calc-rasp.sh output.pdb chainID out_file_name output_directory -
Mol* viewerStructure predictions are visualized in MutationExplorer with an adapted and extended version of Mol* https://doi.org/10.1093/nar/gkab314, the successor of the NGL Viewer https://doi.org/10.1093/nar/gkv402. The extensions of Mol* from MDsrv https://doi.org/10.1093/nar/gkac398 have been incorporated into MutationExplorer so that sequence alignments are integratively displayed near structure visualizations. The adapted Mol* viewer can be found on GitHub.
General remarks
-
Limitations
Currently, MutationExplorer has no potentials targeted for membrane proteins available. Especially for residues facing the membrane, a special score-function and preparation is desired. Other residues, especially those outside of the membrane can be investigated with MutationExplorer without limitations.
Moreover, some mutations are generally challenging for Rosetta, foremost those from or to proline. Mutations from glycine may require conformational adaptation beyond the protocols used here, but proteins may be more flexible.
Ligands can only be handled as far as they are included in Rosetta’s energy/scoring functions.
MutationExplorer cannot handle very large structures, such as CryoEM structures available only in CIF format. Partial AlphaFold models for transcripts with more than 2,700 amino acids are not supported. We also caution that for large structures our default protocols for minimizations might be insufficient. Users should definitely minimize these on their local computer before upload. For this purpose, we recommend using the according Tutorial command lines.
Happy mutating!
Please cite: MutationExplorer: a webserver for mutation of proteins and 3D visualization of energetic impacts