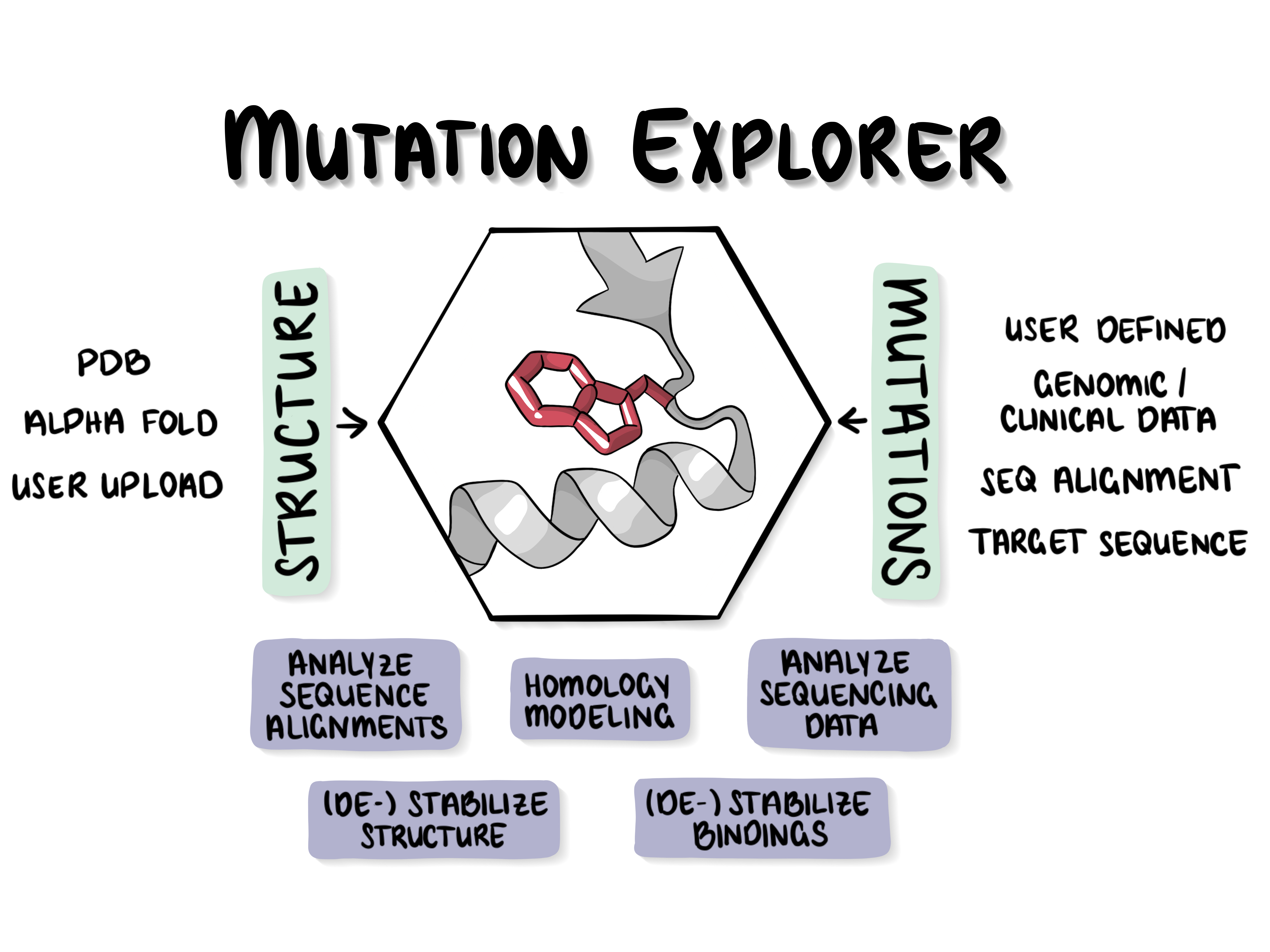
The server lets users mutate proteins in various ways and study mutation effects in 3D. You can evolve a protein over many mutation rounds within the viewer under consideration of the mutational effect on the structure. The only required inputs are a structural model and at least one mutation of interest or a target sequence/alignment. Alternatively, the user can upload human SNPs in the VCF format and obtains a visualization of the (de-)stabilizing effect of those mutations on the protein structure.
Go ahead & try it out! You can easily just try out the server and explore or follow one of our tutorials.
If you don't know what input is expected, you can click on the buttons at each field in the forms to get further information.
Find several tutorials about the usage of the server in our tutorials tab. For more detailed explanation about the methods and the server, incl. interpretation of the results, check out the documentation. Frequently asked questions can be found in the FAQ. If you want to reach out to us, you find the information at the tab "Contacts".
MutationExplorer is freely available to all users without any login requirement.
Please cite: MutationExplorer: a webserver for mutation of proteins and 3D visualization of energetic impacts
-
General philosophy of the MutationExplorer
The MutationExplorer maps variants onto the protein 3D structure which allows to interactively explore the effects of mutations with respect to stability and function. Often wwPDB structures contains mutations and can not intuitively be mapped to its native sequence, sequences of structures changed to close or remote homologs or structures designed interactive while inspecting the structure. With the MutationExplorer this is now easily possible and even visualises the effect of the variant on the protein structures stability.
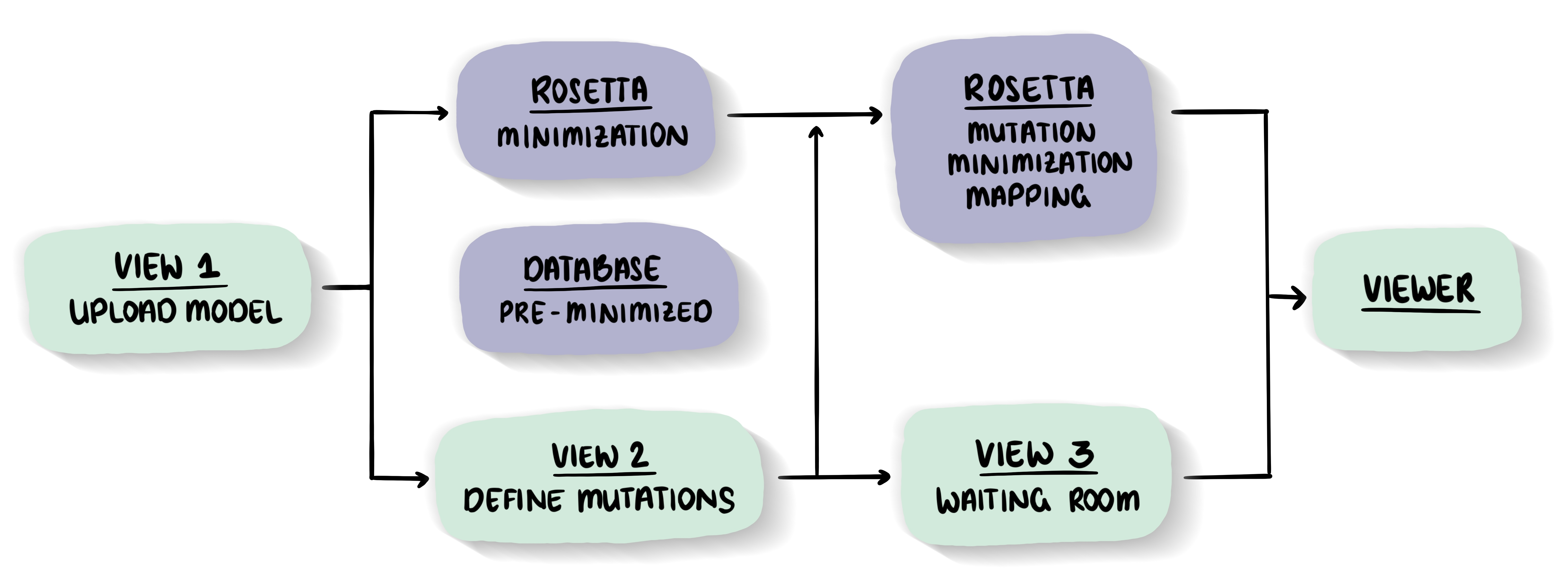
Energy minimization - the key to high qualityThe minimization is crucial for the quality of the outcome. The better the structure is energetically minimized, the more reliable the results will be.
Minimizations via the MutationExplorer
We offer a 'long' and 'short' side-chain only optimization. Find out more in the documentation.Database of pre-minimized structures from the Protein Databank (PDB)
Since the minimization is essential for correctly highlighting the energetic impact of mutations, but takes a substantial amount of time to perform, we calculated a database of pre-minimized structures from the PDB. Currently our database contains 45,000 models. -
Mutational tools - under the the hood of MutationExplorerMutational effects
We use a fast Rosetta fixbb function to calculate the energy difference between wild-type and variant structure. This ΔΔG is displayed in our Explorer. Fin out more in the documentation.
RaSP - bringing exploration to full bloomRaSP is a new deep-learning-based tool that rapidly estimates protein stability changes. RaSP predictions strongly correlate to scores from Rosetta calculations which demand longer compute times, especially if not only one or a few mutations are calculated but a full site-saturation library (SSL). By using this, we can get any mutation at each position of the protein for further mutational consideration and exploration within the viewer.
With RaSP, MutationExplorer presents the user with a quick initial estimation of a mutation’s (de)stabilizing effect, without having to wait for the longer full minimization process.
Include ensembl information via VCF filesUnder the hood, the sequence data will be translated into mutations, an AlphaFold structure will be generated of the associated protein and the variants will be highlighted.
-
MutationExplorer serves many applications - see a selectionHighlights & common applications
Within the tutorials, we will show use-cases of the MutationExplorer, among others
- Visualize human sequencing information and their effect on the protein level.
- Mutation of individual structures from the wwPDB, UniProt or Molecular Dynamics snapshots.
- See the mutational effect when transforming the sequence of a structure into a close or distant homolog via sequence alignment or input sequences.
- Modify your protein-protein interface to increase or decrease binding affinity.
What AlphaFold is not able to do is to model different states of a protein. This can be achieved using MutationExplorer. For many classes and families of proteins, multiple states are available in the PDB. Different states can be modeled By selecting different PDBs as base structure and adding a target sequence or alignment.
Backmutating PDB structures to wildtype sequence, e.g. for dockings and Molecular Dynamics simulationsStructures deposited in the PDB are very often modified in order to solve them experimentally. Using MutationExplorer, it is a simple two-step process to mutate them back to their native sequence. Thus it is common step for simulating proteins to back-mutate PDB.
Multi-state modeling of homologs via Molecular Dynamics simulation ensemblesMolecular dynamics simulation can generate ensembles of functionally relevant states. MutationExplorer maps those ensembles to close or distant homologs. For close homologs, an alignment will be automatically generated internally. Alternatively, a pre-calculated sequence alignment can be provided. Where more refined alignments are required, the server accepts results forwarded from the AlignMe website https://doi.org/10.1093/nar/gkac391 Such sequence alignments carry nuanced details and MutationExplorer drills down to reveal them all.
Aiding design for experimental studies for protein characterizationWhen designing proteins, the effect of the variant is often only known after applying of an external software, via multiple design rounds. After each of those, the structures can be inspected. The power of the MutationExplorer lies in the possibility to do this directly in the viewer and select novels rounds of mutations immediatley and due to their influence on the structure.
(De-) Stabilize a protein-protein interface3D mapping of energies is not limited to monomers. Over single or multiple rounds of mutations, protein complexes or protein-protein interfaces can flexibly be designed to achieve (de)stabilization. The effect of each design choice can be explored visually.
-
General remarksRuntime
Runtime might vary on the protein length, whether the structure has already been minimized or if RaSP has been selected. In general, the pre-process until you reach the visualization page will require the most waiting time. After that, the exploration will not require further waiting time. Find more information in the Documentation or FAQ.
LimitationsA full list of limitations can be found in the documentation. Of importance is that the MutationExplorer at its current state does not take extra care about membrane proteins, hetero atoms such as water, ions or ligands and non-standard amino acids incl. disulfid bridges.
Further, the interpretation of proline and glycine mutations should be handled with care as the selected Rosetta protocol is not suited for the necessary flexibility of those modifications.
The MutationExplorer is its current state not able to complete a homology model by modeling missing parts in the PDB, e.g. when a sequence alignment has been provided.
Finally, the uploaded structure file can not be in CIF format and partial AlphaFold models for transcripts with more than 2,700 amino acids are not supported. We also caution that for large structures our default protocols for minimizations might be insufficient. Users should definitely minimize these on their local computer before upload (see Tutorial).